A transformative framework for analyzing molecular dynamics simulations through advanced clustering algorithms
Installation • The Problem • Our Solution • Key Features • Quick Start • Publications • Contributing
Molecular Dynamics (MD) simulations generate terabytes of conformational data, but extracting meaningful biological insights remains challenging. Traditional clustering methods struggle with:
- Exponential complexity - MD datasets are massive.
- Poor initialization - leading to suboptimal clustering.
- Pathway ambiguity - difficulty identifying dominant biological pathways.
- Native structure prediction - accurately identifying biologically relevant states.
- Pairwise similarity limitations - traditional methods only compare pairs of objects, causing performance bottlenecks.
- Stochastic variability - lack of reproducibility across clustering runs.
MDANCE introduces a novel n-ary similarity framework that transforms how we analyze MD trajectories. Our algorithms provide:
- Linear scaling - from O(N²) to O(N) complexity.
- Deterministic results - reproducible science.
- Biological relevance - algorithms designed for structural biology.
- Unprecedented accuracy - validated against experimental structures.
- Extended similarity techniques - swift identification of high and low-density regions in linear time.
Breakthrough: Deterministic centroid initialization using n-ary comparisons to identify high-density regions and select diverse initial conformations.
Key Advantages:
- Solves the seed selection challenge in k-means clustering.
- Creates compact, well-separated clusters that accurately find metastable states.
- Provides consistent cluster populations across replicates.
- Dramatically reduces runtime: clusters 1.5 million HP35 frames in ~40 minutes.
Breakthrough: Combines k-means efficiency with hierarchical flexibility using n-ary difference functions.
Performance:
- Retains k-means computational efficiency while enabling arbitrary partitions.
- Successfully analyzes simulations with over 1.5 million frames.
- Achieves in ~34 minutes what traditional HAC requires 29 hours for 1.5 million frames.
- Builds hierarchy without expensive pairwise distance matrices.
Breakthrough: Top-down hierarchical clustering framework that recursively splits clusters based on n-ary similarity principles.
Key Features:
- Completely avoids O(N²) pairwise distance matrices.
- Deterministic anchor initialization with NANI.
- Multiple cluster selection criteria including weighted variance metric.
- Single-pass design enables efficient resolution exploration.
- Matches or exceeds bisecting k-means quality with reduced runtime.
Innovation: Adapts BIRCH CF-tree to molecular dynamics data with RMSD-calibrated merge tests.
Key Capabilities:
- Online clustering that processes frames as they arrive.
- Merge test calibrated directly to RMSD for physical interpretability.
- Completely avoids pairwise distance matrices.
- Scales near-linearly with number of frames.
- Two practical protocols: RMSD-anchored runs and blind sweep analysis.
- Processes hundreds of thousands of frames on a single CPU core in seconds.
Transformative: Hierarchical clustering that identifies dominant biological pathways from enhanced sampling data.
Key Advantages:
- Streamlines analysis of pathway ensembles from multiple MD simulations.
- Integrates n-ary similarity with cheminformatics-inspired tools.
- Identifies most representative pathway within each pathway class.
- Provides insight into dominant biomolecular transformation mechanisms.
- Lower computational cost than Fréchet distance approaches.
- Successfully applied to alanine dipeptide and adenylate kinase systems.
Innovation: Transforms O(N²) Radial Threshold Clustering into O(N) algorithm with novel seed selection and tie-breaking.
Key Features:
- Uses k-means++ for efficient seed selection.
- Employs extended similarity indices for deterministic results.
- Eliminates memory-intensive pairwise RMSD matrices.
- Produces compact and well-separated clusters matching RTC quality.
Novelty: Bridges the gap between efficient k-means and robust density-based clustering using n-ary similarity framework.
Key Advantages:
- Swiftly pinpoints high and low-density regions in linear O(N) time.
- Enables focused exploration of rare events.
- Identifies most representative conformations efficiently.
- Overcomes limitations of pairwise similarity operations.
|
Game Changer: Predicts native protein structures from simulation data with unprecedented accuracy. Scientific Validation: PRIME (Protein Retrieval via Integrative Molecular Ensembles) perfectly mapped all structural motifs in benchmark studies and consistently identified native structures within 2Å RMSD of experimental data. |
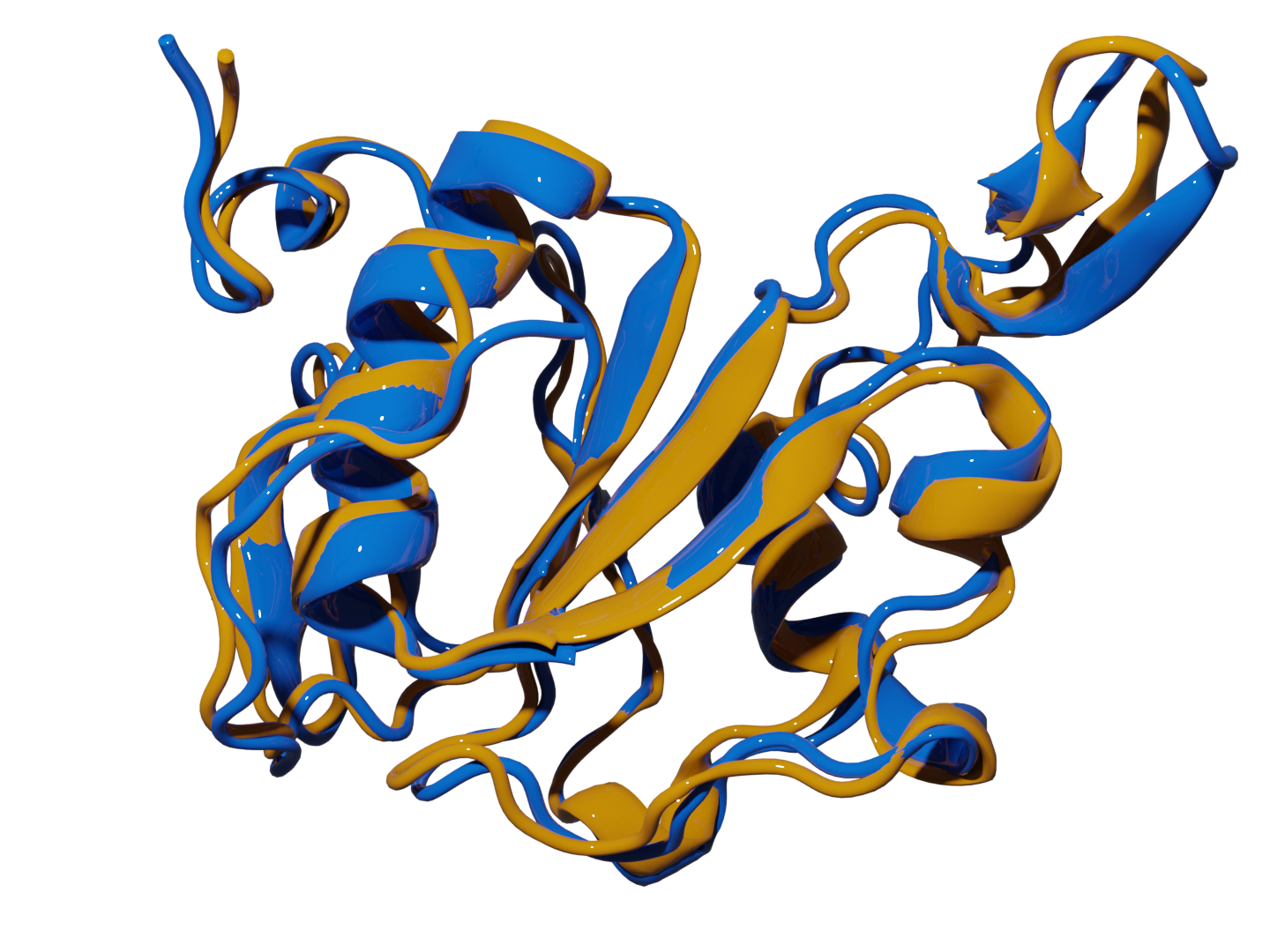 Superposition of native structure using PRIME (yellow) and experimental native structures (blue) of 2k2e.
Superposition of native structure using PRIME (yellow) and experimental native structures (blue) of 2k2e.
|
| Algorithm | Complexity | Type | Key Feature | Best Use Case |
|---|---|---|---|---|
| NANI | O(N) | Initialization | Deterministic centroids | k-means improvement |
| HELM | O(N) | Hybrid hierarchical | k-means + hierarchical fusion | Large-scale analysis |
| DIVINE | O(N) | Divisive hierarchical | Top-down splitting | Multi-resolution analysis |
| mdBIRCH | O(N) | Online clustering | Streaming data processing | Large-scale trajectories |
| SHINE | O(N) | Hierarchical | Pathway analysis | Enhanced sampling |
| eQual | O(N) | Flat clustering | Linear RTC replacement | General purpose |
| CADENCE | O(N) | Density-based | n-ary density estimation | Rare event detection |
| PRIME | O(N) | Post-processing | Native structure prediction | Structure validation |
pip install mdanceimport mdance
import numpy as np
# Load your MD trajectory data
data = np.load('trajectory.npy')
# Use NANI for optimal clustering initialization
from mdance.cluster.nani import KmeansNANI
nani = KmeansNANI(data, n_clusters=5, metric='MSD')
optimal_centroids = nani.initiate_kmeans()
# Cluster with standard *k*-means
from sklearn.cluster import KMeans
kmeans = KMeans(5, init=optimal_centroids[:5], n_init=1)
labels = kmeans.fit_predict(data)- NANI Tutorial - Smart k-means initialization.
- HELM Tutorial - Scalable hierarchical clustering.
- DIVINE Scripts- Deterministic divisive clustering. 1-
run_divine.py, 2-analysis_db.ipynb, 3-assign_labels.py. - mdBIRCH Script - Online clustering for streaming MD data.
- SHINE Script - Pathway analysis.
- eQual Tutorial - Linear-time clustering.
- CADENCE Tutorial - Density-based clustering (to be added).
- PRIME Tutorial - Native structure retrieval.
Our methods are backed by peer-reviewed research:
- NANI: J. Chem. Theory Comput. 2024
- HELM: J. Chem. Inf. Model. 2025
- DIVINE: BioRxiv 2025
- mdBIRCH: BioRxiv 2025
- SHINE: J. Chem. Inf. Model. 2025
- eQual: J. Chem. Inf. Model. 2025
- CADENCE: J. Chem. Inf. Model. 2025
- PRIME: J. Chem. Theory Comput. 2024
MDANCE is enabling researchers to:
- Accelerate drug discovery by rapidly identifying biologically relevant conformations.
- Understand disease mechanisms through precise pathway analysis.
- Validate computational models against experimental structures.
- Scale analyses to massive simulation datasets.
We welcome collaborations and contributions! Whether you're a:
- Computational biologist with novel analysis needs.
- Method developer interested in extending our framework.
- Structural biologist with challenging datasets.
- Open an issue for bug reports or feature requests.
- Submit a pull request for improvements.
- Reach out to discuss research collaborations.
This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM150620.